Hydrogen bonds occur when a "donor" atom donates its covalently
bonded hydrogen atom to an electronegative "acceptor" atom.
The oxygen in -OH (e.g. Ser, Thr, Tyr), HOH, and the nitrogen in -NH3+
(as in Lys, Arg) or
-NH- (as in the main chain peptide bond, Trp, His, Arg, nucleotide bases)
are typical donors.
The lone electron pairs on these same donors can serve as hbond acceptor
sites. So can those on carbonyl oxygens =O (as in the main chain)
or nitrogens with three covalent bonds =N- (as in His, Trp, or nucleotide
bases). Lacking hydrogens, these latter cannot serve as donors.
Jeffrey categorizes hbonds with donor-acceptor
distances of 2.2-2.5 Å as "strong, mostly covalent", 2.5-3.2 Å
as "moderate, mostly electrostatic", 3.2-4.0 Å as "weak, electrostatic" (page 12).
Energies are given as 40-14, 15-4, and <4 kcal/mol respectively.
Most hbonds in proteins are in the moderate category, strong hbonds requiring
moieties or conditions that are rare within proteins. The hydrogen atoms
in moderate hbonds often do not lie on the straight line connecting the
donor to acceptor, so donor-acceptor distance slightly underestimates the
length of the hbond (Jeffrey, p. 14). The mean donor-acceptor distances
in protein secondary structure elements are close to 3.0 Å, as are
those between bases in Watson-Crick pairing (Jeffrey,
pp. 191, 200).
Since many PDB files
lack hydrogen atoms, the presence of an energetically
significant hydrogen bond can be inferred
when a probable donor and acceptor are within 3.5 Å of each other.
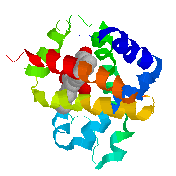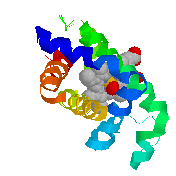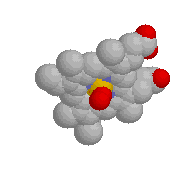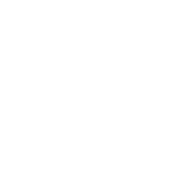
PE's
DISPLAY Contacts defines "likely noncovalently bonded"
oxygens and nitrogens (shown as balls) as those within 3.5 Å of
other oxygens and nitrogens.
At present, PE can display as rods connecting atoms
only two subsets of hydrogen
bonds: protein backbone-to-backbone hbonds within chains (but not between
chains), and Watson-Crick hbonds between DNA base pairs.
These can be shown in QuickViews: DISPLAY Hbonds,
where further information will be shown automatically.
PE presently has no built-in routines to show hbonds between backbone
and sidechain, backbone and water, sidechain and sidechain, sidechain and water,
protein and ligand, protein and nucleic acid,
non-canonical hbonds in DNA or RNA, etc. However, manual methods are available
to show
arbitrary bonds.
 Help, Index & Glossary for
Protein Explorer (PE).
Help, Index & Glossary for
Protein Explorer (PE).


 Animations.
Animations.
 near the
command input slot. There you will find links to the Command
Reference Manuals.
PE simplifies typing commands with its
command aliases.
See also
scripts of commands.
near the
command input slot. There you will find links to the Command
Reference Manuals.
PE simplifies typing commands with its
command aliases.
See also
scripts of commands.
 Control panel
Control panel
 Entering a command.
Entering a command.

 Message Box.
Message Box.

{kind=link}
{kind=link}
{kind=link}